This is one of the first prospective randomised controlled trials applying high dose of intravenous vitamin C (HIVC) to treat COVID-19.
‘High-dose’ vitamin C therapy lacks a universal definition. A previous meta-analysis considered high doses as equal to or greater than 10 g/day. In this trial, we will administer 24 g vitamin C per day for 7 days intravenously.
HIVC has advantages in terms of stability, availability, safety and cost compared with other treatments.
The sample size was calculated in two stages to ensure the calculation is reasonable, maximises the possibility of obtaining significant results and provides credible outcome data.
As the duration and distribution of infected cases are unpredictable geographically and temporally, the number of recruited patients at each centre is also unpredictable, in spite of competitive enrolment.
The COVID-19 pandemic is a threat that has caused panic at the global level. As of 21 June 2020, 8 708 008 cases were confirmed worldwide, resulting in 461 715 deaths.1 Infected patients presented predominantly with fever and cough as well as dyspnoea and myalgia.2–4 A meta-analysis4 concluded that, among the patients, 32.8% presented with acute respiratory distress syndrome (ARDS), 20.3% were transferred to the intensive care unit (ICU) and 13.9% died. According to our previous research,3 patients with ARDS accounted for 66.1% of patients with COVID-19 in the ICU, and the rates of non-invasive and invasive mechanical ventilation were 41.7% and 47.22%, respectively. However, no specific treatment is currently available because traditional antiviral drugs do not work well against COVID-19. Recent clinical trials exploring new therapies, including remdesivir,5 hydroxychloroquine6 and lopinavir-ritonavir,7 for COVID-19 had negative results. Therefore, it is urgent to explore effective therapies considering the grim situation.
The mechanism of COVID-19 involves a cytokine storm, which is a potentially fatal immune reaction triggered by a variety of factors, including infections. Cytokine storms are associated with the clinical manifestation of severe inflammation and highly elevated levels of proinflammatory cytokines.8 Previous research9 suggested that cytokine storms may be the main mechanism of highly pathogenic human coronavirus infected pneumonia, such as severe acute respiratory syndrome and Middle East respiratory syndrome. As a member of the coronavirus family,10 severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) exhibits similar clinical features. The clinical characteristics of COVID-19 indicate that cytokine storms may be positively correlated with the severity of the disease.2 11 12 Moreover, cytokines and T cell subsets may be indicators for predicting prognosis.12 An immunopathology report12 speculated that interleukin (IL)-6 and granulocyte-macrophage colony-stimulating factor (GM-CSF) were the main cytokines in the hyperinflammatory response caused by COVID-19, while Th1 cells were the key cells involved, especially for patients transferred into ICUs. The findings and speculations were verified by an autopsy study that confirmed that patients with critically ill COVID-1913 had developed ARDS. In addition, they also found that the overactivation of T cells and the decrease in the cell counts may explain the occurrence of cytokine storms and, to some degree, the severe immune-medicated injury. All these results indicated that decreasing cytokine storms and immunogenetic damage may be the main treatment option for critically ill patients with COVID-19.
Vitamin C (VC), a common and necessary nutrient, is also an antioxidant. In addition to its role in the metabolism of the human body, including energy transformation, collagen biosynthesis and repair, adrenal steroid and catecholamine production, iron absorption and so on,14–19 VC also possesses antimicrobial properties—thus reducing the risk of infections—and immunomodulatory functions, particularly in high concentrations.14 First, VC plays a crucial role in immunomodulation. It can inhibit the activation of nuclear factor kappa-B (NFκB), which is a primary proinflammatory transcription factor, and plays a pivotal role in overall immunity, including the genetic regulation of chemokines, cytokines, adhesion molecules, inflammatory mediators and apoptosis inhibitors.20 VC can inhibit the production of IL-6 and tumor necrosis factor alpha (TNF-α),21 22 and this effect appears to be a dose dependent.21 VC can reduce GM-CSF signalling responses,23 functioning as a regulator of cytokine redox-signal transduction in host defence cells and having a possible role in controlling inflammatory responses. In addition, high-dose VC can regulate the proliferation and function of T cells, B cells and natural killer (NK) cells.24–27 This may help inhibit the progression of cytokine storms and improve the host’s immunity. Second, previous studies22 demonstrated that VC can inhibit oxidative stress, an important part of the innate immune response to viral respiratory infection28 29 and contributes to lung injury and barrier dysfunction.28 Oxidative stress may also play a role in the mechanism of COVID-19.29 It has been reported that VC can repair oxidative damage in human bronchial epithelium by modulating reactive oxygen species (ROS) generation and inflammatory expression30 and can prevent ROS-induced lung damage.31 Third, VC can regulate alveolar fluid clearance by enhancing lung epithelial barrier function through epigenetic and transcriptional enhancement of protein channels that regulate alveolar fluid clearance.32 33 VC may help decrease the symptoms of ARDS and improve respiratory function. Fourth, VC may have antiviral effects. VC has been reported to inhibit the replication of herpes simplex virus 1, poliovirus type 1 and influenza A virus in vitro.34
Previous research35 demonstrated that intravenous VC (IVC) can achieve a higher plasma concentration than oral VC due to losses during intestinal absorption, tissue transport and renal reabsorption. The efficacy and safety of intravenous high-dose VC (HIVC) in critically ill patients have been investigated via several clinical trials. A recent meta-analysis,15 of mostly cardiac surgery trials, revealed that VC shortened ICU length of stay and duration of mechanical ventilation in ICU patients. In addition, the effect of VC was significantly greater for patients with more severe illness.15 36 Regarding patients with sepsis or ARDS, a phase I trial of IVC in patients with severe sepsis37 reported that VC significantly reduced multiorgan failure scores and circulating injury biomarker levels. Notably, the effect appeared greater in the high-dose group (200 mg/kg) than in the low-dose group (50 mg/kg). However, another study conducted on patients with sepsis and ARDS38 reported no difference in the primary outcome of organ failure scores and inflammation biomarkers but found a significant reduction in 28-day mortality and long-term prognosis. This study applied 50 mg/kg VC intravenously at a late stage when patients were undergoing mechanical ventilation. In addition, there was also a case report39 that administered HIVC (200 mg/kg) to successfully treat virus-induced ARDS, with patients making a rapid recovery after receiving extracorporeal membrane oxygenation and without any long-term sequelae.
A major concern regarding the use of high-dose VC is its potential side effects. Many reported side effects of high-dose VC are insignificant and rare and of little consequence.40 It was reported that high-dose VC was related to haemolysis in glucose-6-phosphate dehydrogenase (G-6-PD) deficiency, acute kidney injury (AKI) and acute oxalate nephropathy.41–44 However, adverse effects were mostly reported in a few cases and were related to too large of doses,41–44 non-standard administration43 or high-risk underlying diseases.44 Clinical trials with large sample sizes conducted on ICU patients15 36–38 reported few adverse events (AEs). In addition, a survey45 also indicated that other than the known complications of kidney damage and G-6-PD deficiency, HIVC appeared to be remarkably safe. For patients with haemochromatosis, G-6-PD deficiency, renal dysfunction, renal stones or oxaluria, VC should be carefully administered; adequate hydration, appropriate dilution and slow infusion rates are recommended for HIVC.46
Thus, we hypothesise that administering HIVC at an early stage of ARDS in COVID-19 would result in better outcomes. In this trial, we will tentatively explore the safety and efficacy of HIVC used in COVID-19.
The main goal is to investigate a new potential therapy for COVID-19 by clarifying the effect of HIVC on the prognosis of patients with COVID-19, especially on respiratory function assessed by ventilation-free days.
This protocol was written in accordance with the Standard Protocol Items: Recommendations for Interventional Trials guidelines47 (see attached Research Checklist); the protocol is summarised in figures 1 and 2.

Standard Protocol Items: Recommendations for Interventional Trials checklist. ALT, alanine aminotransferase; AST, aspartate aminotransferase; BNP, brain natriuretic peptide; BUN, blood urea nitrogen; CHOL, cholesterol; CK-MB, creatine kinase isoenzyme-MB; cysC, cystatin C; DBIL, direct bilirubin; IL-6, interleukin-6; MYO, myoglobin; PCT, procalcitonin; SCR, serum creatinine; TG, triglyceride; TBIL, total bilirubin; TnIU1tr, high sensitive troponin I. 1Markers of myocardial injury include CK-MB, MYO, TnIU1tr. 2Serum chemistry includes ALT, AST, TBIL, DBIL, CHOL, TG, Scr, BUN, CysC, BNP, PCT, cytokines.
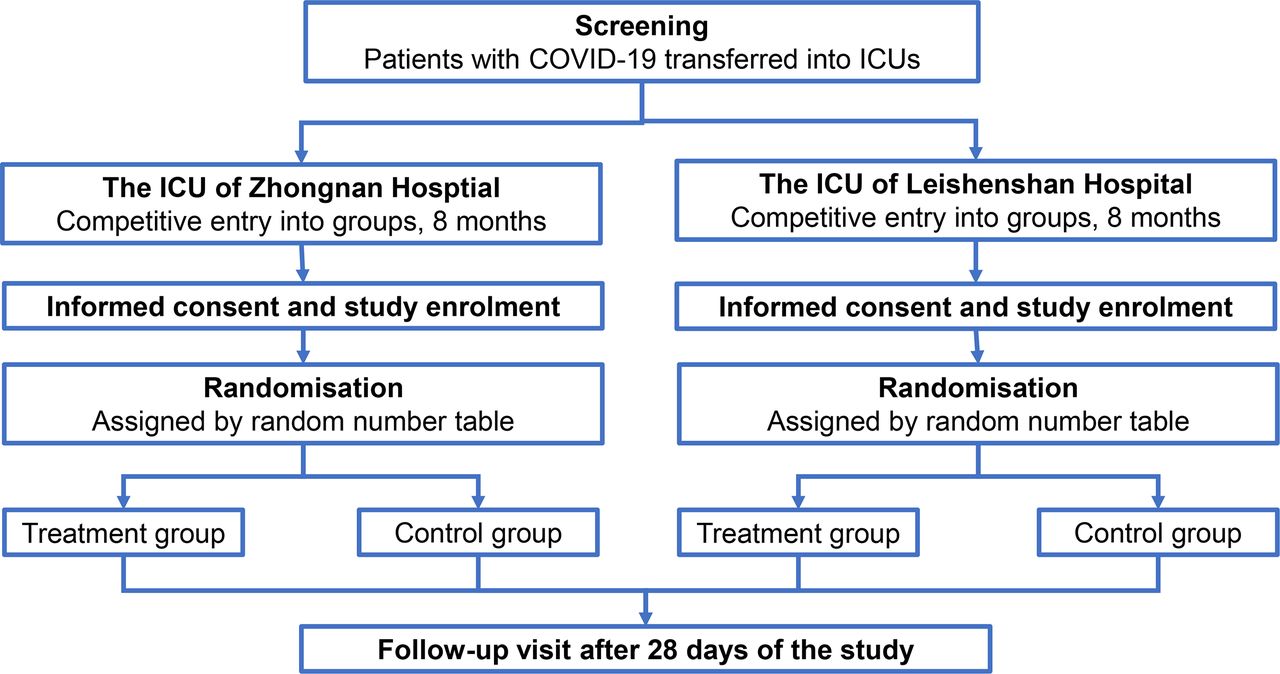
Study flow. ICU, intensive care unit.
The study is a multicentre, prospective randomised, placebo-controlled trial. It is an interventional study with two arms (VC group and placebo group). The study is well designed according to the concept of a pragmatic trial48 with broad inclusion criteria, a reasonable sample size and study procedures embedded into clinical routine care and executed by clinical personnel.
The trial is being conducted in the ICU of Zhongnan Hospital of Wuhan University and the ICU of Leishenshan Hospital of Wuhan City from 14 February 2020 to 30 September 2020. Zhongnan Hospital of Wuhan University, with a comprehensive 66-bed ICU, is a tertiary hospital integrating clinical care, scientific research and teaching. During the COVID-19 pandemic, it took charge of Leishenshan Hospital of Wuhan City (the second specialised hospital that was built by Wuhan government within half a month to only accommodate and treat patients with COVID-19). Each ICU will enrol patients competitively for 8 months.
Patients admitted into the above 2 ICUs, consistent with the following inclusion criteria and no exclusion criteria at the time of randomisation, will be considered for enrolment. Enrolment must be completed within 12 hours after patients are admitted to the ICU.
Participants’ inclusion criteria are as follows:
Adults (age 18 years or older).
Diagnosed with COVID-19 according to the Diagnosis and Clinical Management of 2019 novel coronavirus infected pneumonia (trial version 5, revised version).49
Having a respiratory failure index (RIF) (arterial oxygen tension (or pressure)/fractional inspired oxygen) <300 mm Hg.
Being treated in the ICU.
Exclusion criteria are as follows:
Patients with a history of allergy to VC.
Patients with dyspnoea caused by cardiogenic pulmonary oedema.
Pregnant or lactating women.
Patients with an expected survival duration of less than 24 hours.
Patients with a previous history of end-stage pulmonary disease, end-stage malignant tumour, G-6-PD deficiency, diabetic ketoacidosis, active kidney stone disease and severe kidney diseases.
Patients who are already enrolled in another clinical trial.
As mentioned above, this trial is considered to be safe. First, VC is an essential nutrient for the human body, and IVC has been widely used in clinical practice for over five decades.50 Second, patients with greater potential risks of side effects were excluded based on the exclusion criteria. Third, the rate of infusion is as slow as 12 mL/hour. However, AEs and serious adverse events (SAEs) must be observed and followed in accordance with the good clinical practice guidelines issued by the National Medical Products Administration of the People’s Republic of China.51 An AE refers to any untoward medical event that occurs after a human subject receives a drug. SAEs include prolonged hospital length of stay, disability, death and so on. AEs and SAEs will be recorded during the 28 days of observation from enrolment. Either may occur during a subject’s participation in the research and do not need to have a causal relationship with the treatment. Investigators will evaluate the relationship between the events and the intervention and report it to the ethical committee and data and safety monitoring board (DSMB). Benefits and potential risks are written in the informed consent document. Patients will be informed of the purpose, intervention, benefits and possible risks of the study.
Each ICU is assigned an independent, random numeric table that is generated by the primary investigator alone through Microsoft Excel 2019 and carried out by other investigators. Each table has equal numbers of 1 and 2, which represent the placebo group (sterile water for injection) and treatment group (VC), respectively. Once generated, the list will remain unchanged and be kept by the primary investigator who does not participate in the treatment of patients. When a patient is transferred to the ICU and meets the enrolment criteria, the clinician on duty will inform the other investigators and obtain a number from the list according to the chronological order of ICU recruitment. Then, participants are enrolled in the corresponding group. This trial is not completely blinded. The COVID-19 pandemic is a public health emergency that overwhelms the medical system. Thus, there is not enough manpower and material resources to implement the double-blind method, which requires modifying the medical orders system, connecting the ward and the pharmacy and assigning a special nurse to configure and label infusions. Therefore, the grouping and intervention are unknown to the participants and investigators responsible for data collection and statistical analysis but are known to the clinical staff and investigators responsible for recruiting participants. VC injection and sterile water for injection are both colourless and transparent liquids and will be contained in the same brown syringes with different marks and without explanations on the syringe to make sure that patients cannot distinguish the treatment they receive. Investigators, clinicians and DSMB will monitor the whole process of treatment and determine whether to continue the trial if potential AEs occur or if it is in the participants’ best interests to stop treatment, so there is no need for unblinding of the study grouping.
This trial is an interventional study with two arms. The intervention lasted for 7 days. For the treatment group, 12 g VC will be diluted in sterile water to a total volume of 50 mL and will be infused within 4 hours by an infusion pump. This treatment will be repeated every 12 hours. Thus, the total dosage of VC for the treatment group is 24 g per day. The control group is assigned a placebo (50 mL sterile water); the method and speed of administration are identical to those of the treatment group. Sterile water is used for dilution fluid as well as placebo fluid, as the combination of VC and saline has been reported to irritate the vein.52 No other medications or therapeutic schedules other than standard critical supportive care will be applied in this study. The intervention should be commenced within 12 hours after enrolment. Delivery of the assigned VC injection or placebo injection is achieved through the medical orders system, pharmacy supply, and clinical routine in hospitals. VC injection is a common and basic drug and has been widely used in the hospital, so there will not be problems of inventory and logistics.
Baseline data including demographic characteristics, main diagnosis, assigned group, changes in Acute Physiology and Chronic Health Evaluation (APACHE) II scores, Sequential Organs Failure Assessment (SOFA) scores and multiorgan function, especially respiratory function indices, will be collected on the first day. General vital signs, chest radiography and CT scans, respiratory and cardiac ultrasounds, respiratory indices (RIF, parameters of ventilation and so on), infection indices (white cell count, neutrophil count, lymphocyte count, procalcitonin (PCT) and C reactive protein (CRP)), cytokine tests (IL-6, IL-10 and so on), immunological indices (lymphocyte subpopulation), organ function indices (cardiac function, hepatic function, renal function and coagulation function) and blood gas analyses will be collected on the first, third and seventh days. Participants are scheduled for a follow-up visit on the 28th day to track their long-term prognosis and sequelae.
All the data will be collected from the clinical information system of the hospital and recorded in the case report format (CRF). Every participant will be distinguished with a study identifier and initial without full name. An electronic password-protected document containing all the information from the CRFs will be set up for statistical analysis. The analysis process will be performed by the primary investigator and designated teammates who are experienced with trial CRFs. Relevant documents will be stored at the Department of Critical Care Medicine of Zhongnan Hospital of Wuhan University after the study. The confidentiality and safety of participants’ data are guaranteed. All the data will be reserved for 10 years for the purpose of further analysis and investigation.
The primary outcome of this study is ventilator-free days in the 28 days since admission to the ICU.
Secondary outcomes include the following:
ICU mortality, 28-day mortality.
Changes in SOFA scores.
Changes in plasma biomarkers of inflammation: including white cell counts, neutrophil counts/percentage, lymphocyte counts/percentage, PCT, CRP and cytokines (IL-6, IL-10 and so on).
Changes in pulmonary infection estimated by chest radiography/CT imaging and lung ultrasound and changes in pulmonary function measured by Murray lung injury scores.
Rate of organ failure including ARDS diagnosed by the Berlin definition,53 AKI diagnosed by the Kidney Disease Improving Global Outcomes criteria,54 coagulation disorders, immune dysfunction characterised by changes in lymphocytes and so on.
Other clinical outcomes including ventilation days, vasopressor days, ICU length of stay and hospital length of stay to day 28.
Since the COVID-19 pandemic is uncertain and unpredictable, the sample size is calculated in two stages, which is set to adjust statistical power and determine whether there is a promising trend. At the first stage, as no data from COVID-19 regarding our primary outcome are available, ICU mortality (13.57% reported by the WHO on 12 February55) was chosen instead for the calculation of sample size through a non-inferiority test to maximise the possibility of significant results. With an allowable error of 10%, a total of 308 study participants (154 in each group) would result in a power of 80% with a one-sided type 1 error rate (α) of 2.5%, allowing a 10% withdrawal rate in each group. Of course, this calculated sample size is not accurate and needs revision based on the real situation. Thus, we arranged a second-stage calculation. After 50 patients are enrolled and have completed follow-up, interim analysis will be conducted at the second stage, and the required sample size will be recalculated. During interim analysis, the data of these 50 patients will be used and processed according to the following methods. The results of the analysis will be reported to the DSMB.
Measurement data will be described as the mean with SD or median with IQR according to its distribution. Count data will be represented as frequencies and proportions. For univariate analysis, the difference in measurement data will be compared between the VC and placebo groups using t-test/Mann-Whitney U test. χ2 test and Fisher’s exact test will be used for rate comparisons. A mixed linear model will be used to assess the effect of treatment on outcomes and to fit a repeated-measures analysis of variance. Kaplan-Meier analysis with the Wilcoxon test will be used to show the effect of VC on patient survival probability. The testing will be two sided, and a p value <0.05 will be considered statistically significant. SPSS V.22.0 and GraphPad Prism V.6.00 will be used to complete data processing and statistical analysis.
To describe the patients’ baseline characteristics, ensure no differences in distribution and eliminate the confounding factors, the primary analysis will be an intention-to-treat comparison of the general characteristics (sex, age, race, group assignment, principle diagnosis and so on) and other random effects. The secondary analysis will be a comparison of the primary outcome and secondary outcomes between the treatment group and the control group. The statistical methods mentioned above will be applied according to the characteristics and distribution of the data.
If the number of enrolled patients permits (no fewer than five patients in each subgroup of interest), stratified analyses will be carried out in the following subgroups to further clarify the suitable population for administering HIVC:
Patients without ventilation and patients with mechanical ventilation or with a ventilation requirement within 24 hours after admission to the ICU.
Patients with high (above median) or low (below median) Murray lung injury scores at the time of randomisation.
Patients with high (above median) or low (below median) APACHE II scores at the time of randomisation.
Patients with different severities of ARDS, stratified into mild, moderate and severe groups based on RIF.
Patients receiving or not receiving renal replacement therapy.
For every patient, the days of mechanical ventilation and ventilator parameters will be recorded in the medical record system. Thus, data on the primary outcome is very unlikely to be missing. Secondary outcome data might be missing due to record-keeping errors. Multiple imputation methods, including mean completer, hot-deck imputation and regression, will be used to minimise the effect of missing data as much as possible. The data of patients with complete outcomes after conservative imputation will be included in the comparison between the treatment and control groups.
An independent DSMB comprised of two academic intensivists outside the study who are experienced in conducting clinical trials in critical illness is monitoring the progress and safety of the trial. The DSMB is able to pause the trial to investigate or give suggestions on potential safety issues to improve the study design and implementation.
Previous reports revealed that cytokine storms, which can be suppressed by VC, are believed to be the main mechanism in the deterioration of patients with COVID-19.9 11–13 VC, traditionally considered a dietary supplement, has antimicrobial and immunomodulatory properties. HIVC has been proven to be safe and therapeutic in critical care medicine, primarily as an adjunct to the treatment of septic shock and multiple organ failure, where it has been shown to improve outcomes and reduce mortality.15 36–39 ‘High-dose’ VC therapy lacks a universal definition. A previous meta-analysis56 considered high doses as equal to or greater than 10 g/day. Twenty-four grams of VC in this trial is higher than the dosage of VC in previous clinical trials conducted on critically ill patients with severe infection.37 38
To our knowledge, this is one of the first prospective randomised controlled studies of high-dose intravenous VC treatment for COVID-19. Specific treatments for COVID-19 are not available at present. There are several clinical trials exploring immunotherapy.10 Documenting efficacy for any of these approaches requires time, adequate patient numbers and careful analysis. Vaccine development will require even more time for both safety and efficacy testing. Although blood plasma treatment for critical patients with COVID-19 was recommended by the National and Health Commission of the People’s Republic of China, plasma from recovered patients is a rare resource. Compared with these therapies, HIVC has great advantages in terms of stability, availability, safety and cost. Therefore, this trial is highly meaningful as it could potentially save lives at a low price. HIVC is expected to improve pulmonary function and reduce mortality for patients with COVID-19. Furthermore, it may also provide a basis and reference for HIVC treatment of other diseases with similar mechanisms.
However, there are also limitations to our study design and uncertainties during implementation. First, as SARS-CoV-2 is a novel coronavirus, the duration and distribution of infected cases are unpredictable geographically and temporally, so the number of recruited patients at some clinical centres may be low. Thus, competitive enrolment and multiple clinical trial centres are needed to ensure an adequate number of subjects. Second, complete blinding is not feasible due to the lack of available resources, such as placebos in the same package as VC. The study is unblinded for dosing nurses, attending physicians and investigators in charge of enrolling participants, but blinding will be maintained for patients and all other members of the clinical and research team, such as statistical staff, to minimise bias. Third, some patients may have received non-specific, tentative and explorative symptomatic treatment, such as interferon and traditional Chinese medicine, as there is no effective and standardised guideline for treating COVID-19 at the early stage, despite the National and Health Commission recommendations for the diagnosis and management of COVID-19,49 which continues to be updated. These therapies are expected to be effective, but they may act as confounding factors for the trial, and it must be taken into account for data analysis.
Patient recruitment started on 14 February 2020 and will be completed on 30 September 2020.
This trial is conducted in two hospitals: Zhongnan Hospital of Wuhan University and Leishenshan Hospital of Wuhan City. The authors would like to thank the patients and their family members and the nurses, pharmacy staff, fellows and clinicians of the intensive care units at the above hospitals for making this study possible. We would also like to express our gratitude to the medical assistance teams for their help and sacrifice for Wuhan.
E-mail:
Tel: +44 (0) 141 946 6482
Address: Healthcare Skills Training International Ltd
West of Scotland Science Park
Block 7, Kelvin Campus
Glasgow G20 0SP


